刚刚发布的《仿制药质量和疗效一致性评价品种分类指导意见》(2017年第49号)》可谓掷地有声,其目的无疑是推动国内企业尽快进行一致性评价,也意味着仿制药格局将利好于产品质量优先的企业。
“非289品种”竞争已经开始
2016年11月29日,CFDA发布《公开征求仿制药质量和疗效一致性评价品种分类的指导意见的意见》(下称“征求意见稿”),时隔4个月在2017年4月5日终于发布《仿制药质量和疗效一致性评价品种分类指导意见》的通告(2017年第49号)》(下称“最终通告”)。
相对于“征求意见稿”只针对2018年年底以前完成一致性评价的“289品种”的分类做限定,“最终通告”则是针对所有化学药仿制药的分类。这表明《仿制药质量和疗效一致性评价品种分类指导意见》具备普遍性。也暗示“非289品种”虽然不是国家现阶段重点关注产品,但从国家层面而言,总局鼓励各企业开始所有产品的仿制药一致性评价。
由此,为了占据竞争先机,有志于抢夺市场的潜力较大的“非289品种”的厂家,就别再观望了。事实上,已经有不少企业正悄然开展一致性评价。咸达数据V3.2发现,BE备案制启动后,目前在临床试验数据公开启动临床试验的登记号有58条。BE临床启动仅仅是BE备案产品的一部分,能否启动临床与BE临床机构承接项目数量有一定相关性,鉴于目前愿意承接项目的BE临床机构已经全满的状态,预计实际BE备案的数量将多于目前公开的数据。
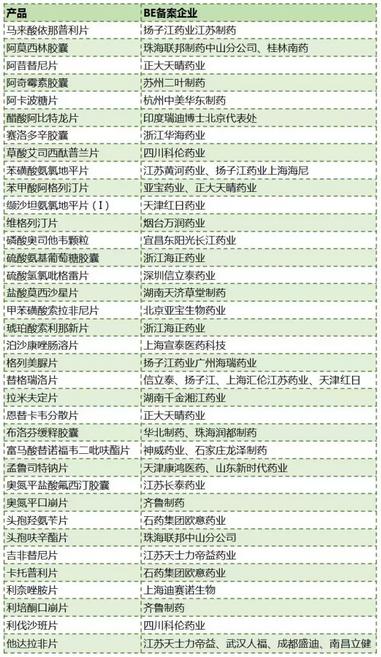
从表1可见,BE备案后启动临床竞争最激烈的产品暂时是替格瑞洛片和他达拉非片。“289目录”中已有BE临床试验启动的产品11个,只占总品种数的3.8%。这11个品种分别是拉米夫定片、阿卡波糖片、硫酸氢氯吡格雷片、头孢呋辛酯片、布洛芬缓释胶囊、马来酸依那普利片、苯磺酸氨氯地平片、格列美脲片、阿奇霉素胶囊、阿莫西林胶囊和卡托普利片。
表1 目前已启动临床的BE备案产品
(数据来源:咸达数据V3.2)
“解放”还是“紧箍”?
“最终通告”对于原研企业在中国境内生产上市的品种能否成为一致性评价的参比制剂描述为,“原研企业在中国境内生产上市的品种,经国家食品药品监督管理总局审核确定发布后,可选择为参比制剂”。
从中检院2017年3月31日所公开的企业参比制剂备案情况的信息可见,已有不少原研药企业申报地产化为参比制剂。例如拜耳拟定阿卡波糖片的參比制剂信息生产厂产地为北京市,氨酚伪麻美芬片Ⅱ、氨麻苯美片则在启东市。赛诺菲将已在北京地产化的格列美脲片申报为参比制剂。强生则将上海市地产化布洛芬混悬液申报为参比制剂。
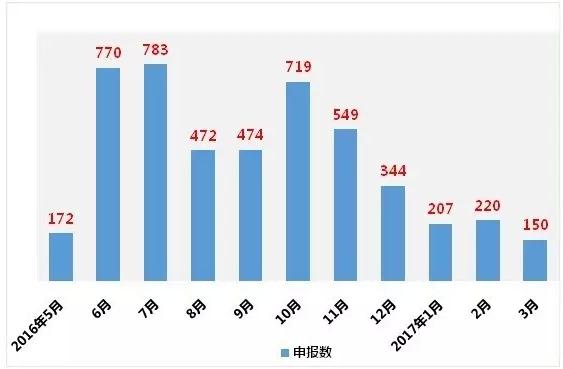
此外,2016年10月之后,企业参比制剂申报数量到达小高峰后一直下降。目前总申报数为4860个,其中“289品种”相关的申报数2648个。289个品种中有235个已申报,占品种数的81%。
图1:2016年5月至2017年3月参比制剂申报数
(数据来源:咸达数据V3.1)
网上不少解读为:这意味着CFDA删去了原研企业在中国境内生产上市的品种自证一致的流程,“审核后可以作为参比,但没有必须都通过自证后成为参比”,对于地产化原研药是一种“解放”。
然而,2017年3月18日总局发布的《普通口服固体制剂参比制剂选择和确定等3个技术指导原则》提到,“若原研企业能证明其地产化药品与原研药品一致,地产化药品也可作为参比制剂使用”。这意味着自证还是必须的,自证的流程请参照指导原则。
至于国内仿制药和进口仿制药,“最终通告”是上市前按照与原研药品质量和疗效一致原则申报和审评的有望走绿色通道获得一致性评价。这与“征求意见稿”的区别主要在于绿色通道的流程描述的差别,产品能否走绿色通道的关键始终在于该药品历史注册申报记录中有无按“一致原则”申报相关证据。
“最终通告”将“征求意见稿”中的“国内特有、膳食补充、辅助治疗等品种”统称为“国内特有品种”。“征求意见稿”原定对此类产品要求为“由食品药品监管总局发布品种名单,并会同行业协会等机构共同研究评价方法,经专家委员会论证后,另行发布”。这更多是公布制定名单的规则。
“最终通告”规定,“由企业选择可重新开展临床试验证明其安全有效性,并参照《化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)》提交申请,后续审核通过后视同通过一致性评价;企业未选择重新开展临床试验的,国家食品药品监督管理总局对外公布其缺乏有效性数据,不建议使用”。这又一次体现了一致性评价主体为企业的原则。现在关键点在于什么样的产品才算“特有品种”?这个定义在文件中并没有体现。而在没有定义的情况下,企业是不会“自投罗网”去做有效性验证试验。
CFDA管理变革进行时:严格而提效
“最终通告”明确了参比制剂、一致性评价审核结果的发布机构是国家食品
药品监督管理总局。
4月5日,国家食品药品监督管理总局发布了《关于调整部分药品行政审批事项审批程序的决定》及其政策解读。药物临床试验审批决定(含国产和进口)、药品补充申请审批决定(含国产和进口)、进口药品再注册审批决定以往是由国家食品药品监督管理总局作出药品行政审批决定,5月1日以后调整为由国家食品药品监督管理总局药品审评中心以国家食品药品监督管理总局名义作出。这意味着除了需要技术审评的工作,其余大多数的药品审评审批权将集中在国家食品药品监督管理总局药品审评中心。
调整后的药物临床试验审批决定(含国产和进口)、药品补充申请审批决定(含国产和进口)、进口药品再注册审批的时限将按照《
药品注册管理办法》规定的行政审批时限执行,预计对应的审批工作有望加快。
从流程的调整和最近CFDA所发布的公告来看,CFDA仍在为加快审评审批做努力,流程优化不代表CFDA对于审评审批的放松,而更应看作为了达到目标,CFDA愿意从自身出发缩短审评审批时间加快
企业申报的效率。
小结<<<
仿制药质量和疗效一致性评价品种分类的发布,意味着一致性评价的又一重要文件的落地。CFDA无论是推动地产化自证也好,加快公布参比制剂名单也罢,最终目的都是为了推动国内企业尽快进行一致性评价工作。
对于过往按一致原则申报的企业和产品,坚持正义始终会得到回馈,而这本来就是这些在规则不明确时仍然坚持
产品质量优先的企业本该应得的尊重。
 京公网安备 11010602104099号
京公网安备 11010602104099号